Introduction to Package “hyperSpec”
The Main Manual of Package “hyperSpec”
Claudia Beleites1,2,3,4,5, Vilmantas Gegzna, Bryan A. Hanson
- DIA Raman Spectroscopy Group, University of Trieste/Italy (2005–2008)
- Spectroscopy \(\cdot\) Imaging, IPHT, Jena/Germany (2008–2017)
- ÖPV, JKI, Berlin/Germany (2017–2019)
- Arbeitskreis Lebensmittelmikrobiologie und Biotechnologie, Hamburg University, Hamburg/Germany (2019 – 2020)
- Chemometric Consulting and Chemometrix GmbH, Wölfersheim/Germany (since 2016)
chemometrie@beleites.de
2024-05-27
Source:vignettes/hyperSpec.Rmd
hyperSpec.Rmd
Package hyperSpec and its associated
packages offer a range of vignettes intended to assist users in
familiarizing themselves with its functionality.
-
The vignettes are accessible through the following links:
-
Alternatively, if you are offline or prefer accessing the vignettes within R, simply type
browseVignettes("hyperSpec")to obtain a clickable list in a browser window. -
Vignettes from other packages:
-
Example
Workflow for 2D Raman Spectra (
chondrodataset) -
Plotting
hyperSpecobjects withggplot2
-
Example
Workflow for 2D Raman Spectra (
1 Things to Know About hyperSpec
Package hyperSpec is an R package that allows convenient handling of hyperspectral data sets, i.e., data sets combining spectra with further data on a per-spectrum basis.
The spectra can be anything that is recorded over a common discretized axis.
This vignette gives an introduction to basic working techniques using the R package package hyperSpec.
This is done mostly from a spectroscopic point of view: rather than going through the functions provided by package hyperSpec, it is organized by spectroscopic tasks.
1.1 Terms & Notations Used Here
Throughout the documentation of the package, the following terms are used:
- wavelength
- spectral abscissa frequency, typical units are wavenumbers, chemical shift, Raman shift, \(\frac{m}{z}\), etc.
- intensity
- spectral ordinate transmission, absorbance, \(\frac{e^{-}}{s}\), intensity, etc.
- extra data
-
further information/data belonging to each spectrum such as
spatial information (spectral images, maps, or profiles), temporal information (kinetics, time series), concentrations (calibration series), class membership information, etc. Class
hyperSpecobject may contain arbitrary numbers of extra data columns.
In R, slots of an S4 class are accessed by the @ operator.
In this vignette, the notation @xxx will thus mean “slot xxx of an object”.
Likewise, named elements of a list and columns of a data.frame are accessed by the $ operator, and $xxx will be used for “column xxx”, and as an abbreviation for “column xxx of the data.frame in slot data of the object” (the structure of hyperSpec objects is discussed in section 1.2).
1.2 The Structure of hyperSpec Objects
hyperSpec objects are defined using an S4 class. It contains the following slots:
-
@wavelengthcontaining a numeric vector with the wavelength axis of the spectra. -
@dataadata.framewith the spectra and all further information belonging to the spectra. -
@labela list with appropriate labels (particularly for axis annotations).
Most of the data is stored in @data. This data.frame has one special column, $spc. This is the column that actually contains the spectra. The spectra are stored in a matrix inside this column, as illustrated in Figure 1.1.
Even if there are no spectra, $spc must still be present.
It is then a matrix with zero columns.
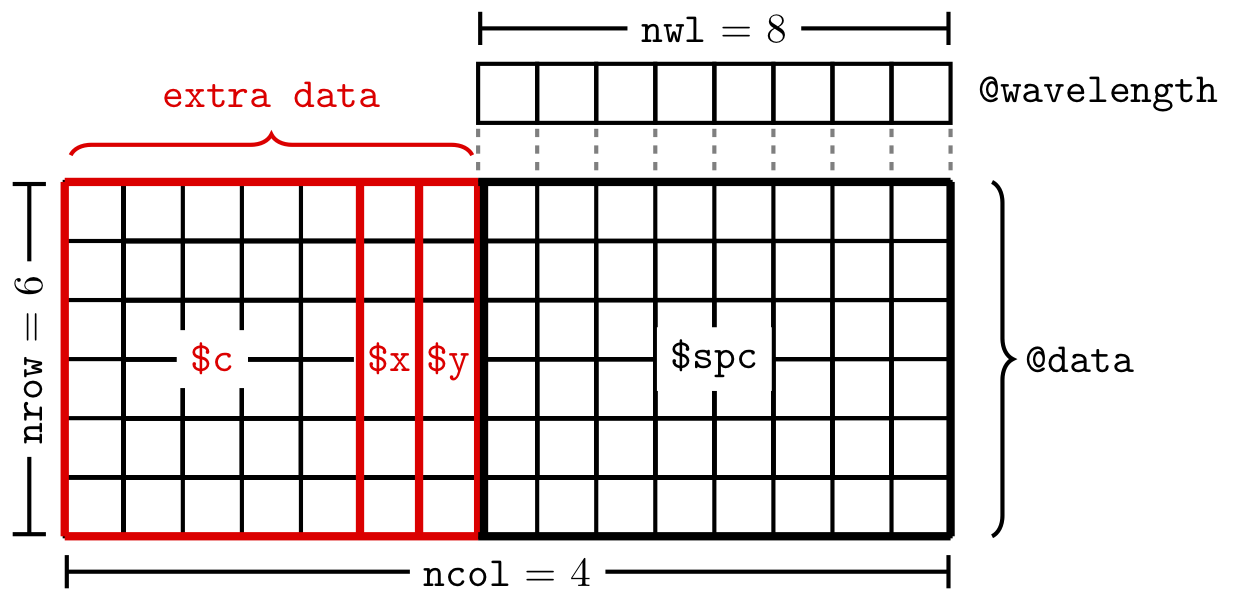
Figure 1.1: The structure of the data in a hyperSpec object. In this example, the ‘extra data’ are the x, y and c columns in @data.
Slot @label contains an element for each of the columns in @data plus one holding the label for the wavelength axis, .wavelength.
They are accessed by their names which must be the same for columns of @data and the list elements.
The elements of the list may be anything suitable for axis annotations, i.e., they should be either character strings or expressions for “pretty” axis annotations (see, e.g., Figure 5.7).
To get familiar with expressions for axis annotation, see ?plotmath and demo(plotmath).
1.3 Data Sets
Package hyperSpec comes with several data sets:
flu |
A set of fluorescence spectra of a calibration series. |
laser |
A time series of an unstable laser emission. |
paracetamol |
A Raman spectrum of paracetamol (acetaminophen) ranging from 100 to 3200 cm-1 with overlapping wavelength ranges. |
faux_cell |
A synthetic data set similar to
chondro. |
barbiturates |
GC-MS spectra with differing wavelength axes as a list of 5 hyperSpec objects. |
In this vignette, the data sets are used to illustrate appropriate procedures for different tasks and different spectra.
In addition, laser and flu are accompanied by their own vignettes showing sample workflows for the respective data type.
This document describes how to accomplish typical tasks in the analysis of spectra.
It does not give a complete reference on particular functions.
It is therefore recommended to look up the methods in the R help system using ?command.
1.4 Options
The global behavior of package hyperSpec can be configured via options.
The values of the options are retrieved with hy_get_options() and hy_get_option(), and changed with hy_set_options().
Table 9.1 gives an overview of the options.
You should not worry about these at the start of your exploration of hyperSpec.
2 Obtaining Basic Information about hyperSpec Objects
Any of print(), show() or summary() provides an overview of the object (the output of each is identical):
faux_cell
#> hyperSpec object
#> 875 spectra
#> 4 data columns
#> 300 data points / spectrumThe data set faux_cell consists of 875 spectra with 300 data points each, and 4 data columns: two for the spatial information, one giving the cell region of each pixel plus $spc.
These details can be directly obtained by
nrow(faux_cell)
#> [1] 875
nwl(faux_cell)
#> [1] 300
ncol(faux_cell)
#> [1] 4
dim(faux_cell)
#> nrow ncol nwl
#> 875 4 300The names of the columns in @data are accessed by colnames().
colnames(faux_cell)
#> [1] "x" "y" "region" "spc"Likewise, rownames() returns the names assigned to the spectra, and dimnames() yields a list of these three vectors (including also the column names of $spc).
The column names of the spectra matrix contain the wavelengths as a character vector, while wl() (see section 3.6.4) returns the numeric vector of wavelengths.
3 Accessing & Manipulating hyperSpec Objects
While the parts of the hyperSpec object can be accessed directly, it is good practice to use the functions provided by the package to handle the objects rather than accessing the slots directly.
This also ensures that proper (i.e. valid) objects are returned.
In some cases, however, direct access to the slots can considerably speed up calculations (see the appendices).
The main functions to retrieve the data of a hyperSpec object are [] and [[]].
The difference between these functions is that [] returns a hyperSpec object, whereas [[]] returns a data.frame containing x$spc, the spectral data.
To modify a hyperSpec object, the corresponding functions are [<- and [[<-. The first form is used to modify the entire hyperSpec object and the second form modifies the spectral data in x$spc.
hyperSpec objects are triple indexed:
x[i, j, l, wl.index = TRUE/FALSE]x[[i, j, l, wl.ndex = TRUE/FALSE]]-
irefers to rows of the@dataslot.ican be integer indices or a logical vector. -
jrefers to columns of the@dataslot.jcan be integer indices, a logical vector or the name of a column. However, there is no guaranteed order tocolnames(x)so using integer indices and logical vectors is unwise. -
lrefers to wavelengths. Note the argumentwl.indexwhich determines howlis interpreted. - If there is only one index given, e.g.
x[1:3], it refers to the row indexi. Likewise, if there are only two indices given they refer toiandj. - See 3.5.1 and 3.4 for even more ways to specify the indices.
3.1 Summary of Extraction Functions for hyperSpec Objects (getters)
Table 3.1 shows the main functions that can be used with class hyperSpec to access the object (“getters”).
| Syntax | Description |
|---|---|
x[] |
Returns the entire hyperSpec object unchanged. |
x[i, , ] |
Returns the hyperSpec object with selected rows; equivalent to x[i]. |
x[, j, ] |
Returns the hyperSpec object with empty x$spc slot. If you want the column j, x[["name"]] returns a data.frame containing j or x$name returns it as a vector. |
x[, , l, wl.index = TRUE/FALSE] |
Returns the hyperSpec object with selected wavelengths. |
x[[]] |
Returns the spectra matrix (x$spc). |
x[[i, , ]] |
Returns the spectra matrix (x$spc) with selected rows. |
x[[, j, ]] |
Returns a data.frame with the selected columns. Safest to give j as a character string. |
x[[, , l, wl.index = TRUE/FALSE]] |
Returns the spectra matrix (x$spc) with selected wavelengths. |
x$name |
Returns the column name as a vector. |
x$. |
Returns the complete data.frame x@data, with the spectra in column $spc. |
x$.. |
Returns all the extra data (x@data without x$spc). |
wl() |
Returns the wavelengths. |
labels() |
Returns the labels. |
One can see that there are several ways to get the spectral data: x$spc, x[[]], x$...
The first two forms return a matrix, while the last returns a data.frame.
3.2 Summary of Replacement Functions for hyperSpec Objects (setters)
Table 3.2 shows the main functions that can be used with class hyperSpec to modify the object (“setters”).
| Syntax | Description |
|---|---|
x[i, ,] <- |
Replaces the specified rows of the @data slot, including x$spc and any extra data columns. Other approaches are probably easier. |
x[, j,] <- |
Replaces the specified columns. Safest to give j as a character string. |
x[i, j] <- |
Replaces the specified column limited to the specified rows. Safest to give j as a character string. |
x[, , l, wl.index = TRUE/FALSE] <- |
Replaces the specified wavelengths. |
x[[i, ,]] <- |
Replaces the specified row of x$spc
|
x[[, j,]] <- |
As [[]] refers to just the spectral data in x$spc, this operation is not valid. See below. |
x[[, , l, wl.index = TRUE/FALSE]] <- |
Replaces the intensity values in x$spc for the specified wavelengths. |
x[[i, , l, wl.index = TRUE/FALSE]] <- |
Replaces the intensity values in x$spc for the specified wavelengths limited to the specified rows. |
x$.. <- |
Sets the extra data (x@data without touching x$spc). The column names must match exactly in this case. |
wl<- |
Sets the wavelength vector. |
labels<- |
Sets the labels. |
For more details on []<-, [[]]<-, and $<-, see 3.5.1 and 3.4.
3.3 Selecting and Deleting Spectra
The extraction function [] takes the spectra (rows) as the first argument (For detailed help: see ?"[").
It may be a vector giving the indices of the spectra to extract (select), a vector with negative indices indicating, which spectra should be deleted, or a vector of logical values.
data(palette_colorblind) # load colorblind-friendly palette
plot(flu, col = palette_colorblind[4])
plot(flu[1:3], add = TRUE)![An example with `[]` operator and positive indices.](hyperSpec_files/figure-html/selspc-1.png)
Figure 3.1: An example with [] operator and positive indices.
![An example with `[]` operator and negative indices.](hyperSpec_files/figure-html/delspc-1.png)
Figure 3.2: An example with [] operator and negative indices.
![An example with `[]` operator and logical indices.](hyperSpec_files/figure-html/selspc2-1.png)
Figure 3.3: An example with [] operator and logical indices.
3.3.1 Random Samples
A random subset of spectra can be conveniently selected by sample():
sample(faux_cell, 3)
#> hyperSpec object
#> 3 spectra
#> 4 data columns
#> 300 data points / spectrumIf indices into the selected spectra are needed instead, use sample(..., index = TRUE):
sample(faux_cell, 3, index = TRUE)
#> [1] 183 797 2793.3.2 Sequences
Sequences of every nth spectrum or the like can be retrieved with seq():
seq(faux_cell, length.out = 3, index = TRUE)
#> [1] 1 438 875
seq(faux_cell, by = 100)
#> hyperSpec object
#> 9 spectra
#> 4 data columns
#> 300 data points / spectrumHere, indices may be requested using index = TRUE.
3.4 Selecting Extra Data Columns
The second argument of the extraction functions [] and [[]] specifies the extra data columns.
They can be given like any column specification for a data.frame, i.e., numeric, logical, or by a vector of the column names. However, since there is intrinsic order in the column names of a hyperSpec object, using the column names is safest:
colnames(faux_cell)
#> [1] "x" "y" "region" "spc"
faux_cell[[1:3, 1]]
#> x
#> 1 -11.55
#> 2 -10.55
#> 3 -9.55
faux_cell[[1:3, -5]]
#> x y region spc.602 spc.606 spc.610 spc.614 spc.618 spc.622 spc.626 spc.630 spc.634
#> 1 -11.55 -4.77 matrix 132 132 150 128 143 113 121 125 121
#> 2 -10.55 -4.77 matrix 72 73 99 84 54 74 68 79 73
#> 3 -9.55 -4.77 matrix 120 132 142 118 125 123 111 146 122
#> spc.638 spc.642 spc.646 spc.650 spc.654 spc.658 spc.662 spc.666 spc.670 spc.674 spc.678 spc.682
#> 1 123 135 139 151 143 133 113 124 104 127 127 131
#> 2 98 83 74 73 69 90 84 80 69 61 65 76
#> 3 123 142 137 141 114 127 120 137 115 127 137 142
#> spc.686 spc.690 spc.694 spc.698 spc.702 spc.706 spc.710 spc.714 spc.718 spc.722 spc.726 spc.730
#> 1 145 134 127 136 121 131 132 131 141 139 109 124
#> 2 80 93 76 80 79 93 81 72 74 76 90 78
#> 3 128 137 118 115 116 126 116 125 111 125 106 123
#> spc.734 spc.738 spc.742 spc.746 spc.750 spc.754 spc.758 spc.762 spc.766 spc.770 spc.774 spc.778
#> 1 136 124 111 127 128 152 133 122 166 132 146 136
#> 2 78 94 86 72 77 76 79 91 69 63 84 109
#> 3 128 119 111 142 139 123 111 138 119 109 126 111
#> spc.782 spc.786 spc.790 spc.794 spc.798 spc.802 spc.806 spc.810 spc.814 spc.818 spc.822 spc.826
#> 1 159 232 267 288 324 312 263 267 216 185 130 150
#> 2 129 160 193 237 260 272 238 203 154 115 104 93
#> 3 172 216 267 282 313 324 280 232 186 182 127 127
#> spc.830 spc.834 spc.838 spc.842 spc.846 spc.850 spc.854 spc.858 spc.862 spc.866 spc.870 spc.874
#> 1 136 156 123 123 131 131 134 128 133 151 143 124
#> 2 98 71 77 66 92 64 84 80 75 79 81 77
#> 3 117 135 123 119 121 125 134 130 125 136 125 135
#> spc.878 spc.882 spc.886 spc.890 spc.894 spc.898 spc.902 spc.906 spc.910 spc.914 spc.918 spc.922
#> 1 132 120 123 117 111 128 117 145 147 116 121 137
#> 2 85 82 85 74 81 76 66 77 74 81 87 85
#> 3 117 144 126 143 148 124 138 131 126 138 110 132
#> spc.926 spc.930 spc.934 spc.938 spc.942 spc.946 spc.950 spc.954 spc.958 spc.962 spc.966 spc.970
#> 1 147 124 137 139 143 128 123 113 120 139 162 108
#> 2 72 91 94 69 75 81 72 84 82 80 70 80
#> 3 135 140 131 123 144 126 118 135 138 144 142 116
#> spc.974 spc.978 spc.982 spc.986 spc.990 spc.994 spc.998 spc.1002 spc.1006 spc.1010 spc.1014
#> 1 141 138 132 145 122 118 105 116 132 122 137
#> 2 86 84 78 80 97 83 66 67 78 90 77
#> 3 146 139 135 134 131 143 135 128 131 139 128
#> spc.1018 spc.1022 spc.1026 spc.1030 spc.1034 spc.1038 spc.1042 spc.1046 spc.1050 spc.1054
#> 1 133 105 114 127 129 142 139 122 114 122
#> 2 89 74 76 82 79 96 83 72 69 86
#> 3 119 126 136 136 138 113 126 150 115 133
#> spc.1058 spc.1062 spc.1066 spc.1070 spc.1074 spc.1078 spc.1082 spc.1086 spc.1090 spc.1094
#> 1 130 136 143 116 147 132 138 137 108 130
#> 2 84 83 95 66 95 96 87 90 80 85
#> 3 126 130 135 133 124 141 135 131 128 141
#> spc.1098 spc.1102 spc.1106 spc.1110 spc.1114 spc.1118 spc.1122 spc.1126 spc.1130 spc.1134
#> 1 154 134 125 155 123 111 132 146 146 122
#> 2 77 88 74 86 76 70 85 71 71 94
#> 3 136 124 136 132 116 148 144 145 147 124
#> spc.1138 spc.1142 spc.1146 spc.1150 spc.1154 spc.1158 spc.1162 spc.1166 spc.1170 spc.1174
#> 1 130 123 122 131 150 144 129 132 133 130
#> 2 96 72 75 86 85 81 87 75 72 82
#> 3 147 137 144 126 124 146 121 155 139 150
#> spc.1178 spc.1182 spc.1186 spc.1190 spc.1194 spc.1198 spc.1202 spc.1206 spc.1210 spc.1214
#> 1 125 118 113 141 131 117 129 136 120 124
#> 2 81 87 82 84 77 80 60 87 81 89
#> 3 103 120 126 135 147 138 124 127 128 124
#> spc.1218 spc.1222 spc.1226 spc.1230 spc.1234 spc.1238 spc.1242 spc.1246 spc.1250 spc.1254
#> 1 152 117 126 126 151 132 134 138 127 117
#> 2 74 80 71 88 77 95 75 78 76 79
#> 3 132 135 151 137 136 132 135 138 136 109
#> spc.1258 spc.1262 spc.1266 spc.1270 spc.1274 spc.1278 spc.1282 spc.1286 spc.1290 spc.1294
#> 1 134 131 122 128 128 153 131 116 143 127
#> 2 81 85 92 83 88 79 88 90 81 87
#> 3 158 143 135 122 129 127 128 135 134 123
#> spc.1298 spc.1302 spc.1306 spc.1310 spc.1314 spc.1318 spc.1322 spc.1326 spc.1330 spc.1334
#> 1 120 125 111 132 144 129 124 128 124 121
#> 2 73 78 91 79 95 96 90 69 87 82
#> 3 140 140 128 134 139 116 123 145 128 134
#> spc.1338 spc.1342 spc.1346 spc.1350 spc.1354 spc.1358 spc.1362 spc.1366 spc.1370 spc.1374
#> 1 123 134 140 136 141 131 149 117 131 134
#> 2 90 101 79 69 63 83 84 73 85 78
#> 3 125 146 149 145 145 129 132 116 126 123
#> spc.1378 spc.1382 spc.1386 spc.1390 spc.1394 spc.1398 spc.1402 spc.1406 spc.1410 spc.1414
#> 1 115 154 127 123 124 120 170 130 138 139
#> 2 77 82 92 92 87 79 77 80 89 75
#> 3 133 136 151 133 165 148 150 111 133 133
#> spc.1418 spc.1422 spc.1426 spc.1430 spc.1434 spc.1438 spc.1442 spc.1446 spc.1450 spc.1454
#> 1 128 125 134 139 157 130 146 131 128 137
#> 2 89 78 100 83 80 91 84 98 82 95
#> 3 125 146 142 143 101 149 130 139 135 143
#> spc.1458 spc.1462 spc.1466 spc.1470 spc.1474 spc.1478 spc.1482 spc.1486 spc.1490 spc.1494
#> 1 142 130 143 121 123 138 148 131 132 125
#> 2 72 95 85 93 84 101 94 95 90 73
#> 3 133 149 144 132 147 150 145 130 122 116
#> spc.1498 spc.1502 spc.1506 spc.1510 spc.1514 spc.1518 spc.1522 spc.1526 spc.1530 spc.1534
#> 1 126 134 112 117 120 145 148 134 131 121
#> 2 84 78 92 70 80 82 78 87 87 105
#> 3 143 122 134 132 149 121 158 143 123 143
#> spc.1538 spc.1542 spc.1546 spc.1550 spc.1554 spc.1558 spc.1562 spc.1566 spc.1570 spc.1574
#> 1 143 116 134 123 132 135 117 130 129 118
#> 2 84 85 76 86 90 87 86 86 95 69
#> 3 160 127 143 149 140 129 145 137 149 138
#> spc.1578 spc.1582 spc.1586 spc.1590 spc.1594 spc.1598 spc.1602 spc.1606 spc.1610 spc.1614
#> 1 122 139 109 115 154 118 124 131 123 148
#> 2 94 97 92 79 90 72 82 81 79 90
#> 3 134 168 154 134 134 153 132 148 129 152
#> spc.1618 spc.1622 spc.1626 spc.1630 spc.1634 spc.1638 spc.1642 spc.1646 spc.1650 spc.1654
#> 1 134 132 132 115 125 119 130 129 128 118
#> 2 90 79 94 84 105 71 74 88 83 74
#> 3 149 147 148 138 166 152 122 144 124 150
#> spc.1658 spc.1662 spc.1666 spc.1670 spc.1674 spc.1678 spc.1682 spc.1686 spc.1690 spc.1694
#> 1 128 143 113 143 128 126 121 126 134 126
#> 2 81 82 104 85 76 98 79 85 108 86
#> 3 128 140 144 127 124 131 143 142 135 134
#> spc.1698 spc.1702 spc.1706 spc.1710 spc.1714 spc.1718 spc.1722 spc.1726 spc.1730 spc.1734
#> 1 151 125 159 130 130 139 127 129 122 138
#> 2 65 75 93 95 94 77 73 82 83 85
#> 3 124 147 133 143 143 127 139 141 169 137
#> spc.1738 spc.1742 spc.1746 spc.1750 spc.1754 spc.1758 spc.1762 spc.1766 spc.1770 spc.1774
#> 1 142 115 128 136 123 124 122 122 129 128
#> 2 69 89 82 87 88 78 103 79 92 85
#> 3 132 153 132 147 155 154 147 143 138 131
#> spc.1778 spc.1782 spc.1786 spc.1790 spc.1794 spc.1798
#> 1 136 133 139 123 119 129
#> 2 66 83 75 87 92 90
#> 3 144 147 143 120 149 154
faux_cell[[1:3, "x"]]
#> x
#> 1 -11.55
#> 2 -10.55
#> 3 -9.55
faux_cell[[1:3, c(FALSE, TRUE)]] # note the recycling!
#> y spc.602 spc.606 spc.610 spc.614 spc.618 spc.622 spc.626 spc.630 spc.634 spc.638 spc.642
#> 1 -4.77 132 132 150 128 143 113 121 125 121 123 135
#> 2 -4.77 72 73 99 84 54 74 68 79 73 98 83
#> 3 -4.77 120 132 142 118 125 123 111 146 122 123 142
#> spc.646 spc.650 spc.654 spc.658 spc.662 spc.666 spc.670 spc.674 spc.678 spc.682 spc.686 spc.690
#> 1 139 151 143 133 113 124 104 127 127 131 145 134
#> 2 74 73 69 90 84 80 69 61 65 76 80 93
#> 3 137 141 114 127 120 137 115 127 137 142 128 137
#> spc.694 spc.698 spc.702 spc.706 spc.710 spc.714 spc.718 spc.722 spc.726 spc.730 spc.734 spc.738
#> 1 127 136 121 131 132 131 141 139 109 124 136 124
#> 2 76 80 79 93 81 72 74 76 90 78 78 94
#> 3 118 115 116 126 116 125 111 125 106 123 128 119
#> spc.742 spc.746 spc.750 spc.754 spc.758 spc.762 spc.766 spc.770 spc.774 spc.778 spc.782 spc.786
#> 1 111 127 128 152 133 122 166 132 146 136 159 232
#> 2 86 72 77 76 79 91 69 63 84 109 129 160
#> 3 111 142 139 123 111 138 119 109 126 111 172 216
#> spc.790 spc.794 spc.798 spc.802 spc.806 spc.810 spc.814 spc.818 spc.822 spc.826 spc.830 spc.834
#> 1 267 288 324 312 263 267 216 185 130 150 136 156
#> 2 193 237 260 272 238 203 154 115 104 93 98 71
#> 3 267 282 313 324 280 232 186 182 127 127 117 135
#> spc.838 spc.842 spc.846 spc.850 spc.854 spc.858 spc.862 spc.866 spc.870 spc.874 spc.878 spc.882
#> 1 123 123 131 131 134 128 133 151 143 124 132 120
#> 2 77 66 92 64 84 80 75 79 81 77 85 82
#> 3 123 119 121 125 134 130 125 136 125 135 117 144
#> spc.886 spc.890 spc.894 spc.898 spc.902 spc.906 spc.910 spc.914 spc.918 spc.922 spc.926 spc.930
#> 1 123 117 111 128 117 145 147 116 121 137 147 124
#> 2 85 74 81 76 66 77 74 81 87 85 72 91
#> 3 126 143 148 124 138 131 126 138 110 132 135 140
#> spc.934 spc.938 spc.942 spc.946 spc.950 spc.954 spc.958 spc.962 spc.966 spc.970 spc.974 spc.978
#> 1 137 139 143 128 123 113 120 139 162 108 141 138
#> 2 94 69 75 81 72 84 82 80 70 80 86 84
#> 3 131 123 144 126 118 135 138 144 142 116 146 139
#> spc.982 spc.986 spc.990 spc.994 spc.998 spc.1002 spc.1006 spc.1010 spc.1014 spc.1018 spc.1022
#> 1 132 145 122 118 105 116 132 122 137 133 105
#> 2 78 80 97 83 66 67 78 90 77 89 74
#> 3 135 134 131 143 135 128 131 139 128 119 126
#> spc.1026 spc.1030 spc.1034 spc.1038 spc.1042 spc.1046 spc.1050 spc.1054 spc.1058 spc.1062
#> 1 114 127 129 142 139 122 114 122 130 136
#> 2 76 82 79 96 83 72 69 86 84 83
#> 3 136 136 138 113 126 150 115 133 126 130
#> spc.1066 spc.1070 spc.1074 spc.1078 spc.1082 spc.1086 spc.1090 spc.1094 spc.1098 spc.1102
#> 1 143 116 147 132 138 137 108 130 154 134
#> 2 95 66 95 96 87 90 80 85 77 88
#> 3 135 133 124 141 135 131 128 141 136 124
#> spc.1106 spc.1110 spc.1114 spc.1118 spc.1122 spc.1126 spc.1130 spc.1134 spc.1138 spc.1142
#> 1 125 155 123 111 132 146 146 122 130 123
#> 2 74 86 76 70 85 71 71 94 96 72
#> 3 136 132 116 148 144 145 147 124 147 137
#> spc.1146 spc.1150 spc.1154 spc.1158 spc.1162 spc.1166 spc.1170 spc.1174 spc.1178 spc.1182
#> 1 122 131 150 144 129 132 133 130 125 118
#> 2 75 86 85 81 87 75 72 82 81 87
#> 3 144 126 124 146 121 155 139 150 103 120
#> spc.1186 spc.1190 spc.1194 spc.1198 spc.1202 spc.1206 spc.1210 spc.1214 spc.1218 spc.1222
#> 1 113 141 131 117 129 136 120 124 152 117
#> 2 82 84 77 80 60 87 81 89 74 80
#> 3 126 135 147 138 124 127 128 124 132 135
#> spc.1226 spc.1230 spc.1234 spc.1238 spc.1242 spc.1246 spc.1250 spc.1254 spc.1258 spc.1262
#> 1 126 126 151 132 134 138 127 117 134 131
#> 2 71 88 77 95 75 78 76 79 81 85
#> 3 151 137 136 132 135 138 136 109 158 143
#> spc.1266 spc.1270 spc.1274 spc.1278 spc.1282 spc.1286 spc.1290 spc.1294 spc.1298 spc.1302
#> 1 122 128 128 153 131 116 143 127 120 125
#> 2 92 83 88 79 88 90 81 87 73 78
#> 3 135 122 129 127 128 135 134 123 140 140
#> spc.1306 spc.1310 spc.1314 spc.1318 spc.1322 spc.1326 spc.1330 spc.1334 spc.1338 spc.1342
#> 1 111 132 144 129 124 128 124 121 123 134
#> 2 91 79 95 96 90 69 87 82 90 101
#> 3 128 134 139 116 123 145 128 134 125 146
#> spc.1346 spc.1350 spc.1354 spc.1358 spc.1362 spc.1366 spc.1370 spc.1374 spc.1378 spc.1382
#> 1 140 136 141 131 149 117 131 134 115 154
#> 2 79 69 63 83 84 73 85 78 77 82
#> 3 149 145 145 129 132 116 126 123 133 136
#> spc.1386 spc.1390 spc.1394 spc.1398 spc.1402 spc.1406 spc.1410 spc.1414 spc.1418 spc.1422
#> 1 127 123 124 120 170 130 138 139 128 125
#> 2 92 92 87 79 77 80 89 75 89 78
#> 3 151 133 165 148 150 111 133 133 125 146
#> spc.1426 spc.1430 spc.1434 spc.1438 spc.1442 spc.1446 spc.1450 spc.1454 spc.1458 spc.1462
#> 1 134 139 157 130 146 131 128 137 142 130
#> 2 100 83 80 91 84 98 82 95 72 95
#> 3 142 143 101 149 130 139 135 143 133 149
#> spc.1466 spc.1470 spc.1474 spc.1478 spc.1482 spc.1486 spc.1490 spc.1494 spc.1498 spc.1502
#> 1 143 121 123 138 148 131 132 125 126 134
#> 2 85 93 84 101 94 95 90 73 84 78
#> 3 144 132 147 150 145 130 122 116 143 122
#> spc.1506 spc.1510 spc.1514 spc.1518 spc.1522 spc.1526 spc.1530 spc.1534 spc.1538 spc.1542
#> 1 112 117 120 145 148 134 131 121 143 116
#> 2 92 70 80 82 78 87 87 105 84 85
#> 3 134 132 149 121 158 143 123 143 160 127
#> spc.1546 spc.1550 spc.1554 spc.1558 spc.1562 spc.1566 spc.1570 spc.1574 spc.1578 spc.1582
#> 1 134 123 132 135 117 130 129 118 122 139
#> 2 76 86 90 87 86 86 95 69 94 97
#> 3 143 149 140 129 145 137 149 138 134 168
#> spc.1586 spc.1590 spc.1594 spc.1598 spc.1602 spc.1606 spc.1610 spc.1614 spc.1618 spc.1622
#> 1 109 115 154 118 124 131 123 148 134 132
#> 2 92 79 90 72 82 81 79 90 90 79
#> 3 154 134 134 153 132 148 129 152 149 147
#> spc.1626 spc.1630 spc.1634 spc.1638 spc.1642 spc.1646 spc.1650 spc.1654 spc.1658 spc.1662
#> 1 132 115 125 119 130 129 128 118 128 143
#> 2 94 84 105 71 74 88 83 74 81 82
#> 3 148 138 166 152 122 144 124 150 128 140
#> spc.1666 spc.1670 spc.1674 spc.1678 spc.1682 spc.1686 spc.1690 spc.1694 spc.1698 spc.1702
#> 1 113 143 128 126 121 126 134 126 151 125
#> 2 104 85 76 98 79 85 108 86 65 75
#> 3 144 127 124 131 143 142 135 134 124 147
#> spc.1706 spc.1710 spc.1714 spc.1718 spc.1722 spc.1726 spc.1730 spc.1734 spc.1738 spc.1742
#> 1 159 130 130 139 127 129 122 138 142 115
#> 2 93 95 94 77 73 82 83 85 69 89
#> 3 133 143 143 127 139 141 169 137 132 153
#> spc.1746 spc.1750 spc.1754 spc.1758 spc.1762 spc.1766 spc.1770 spc.1774 spc.1778 spc.1782
#> 1 128 136 123 124 122 122 129 128 136 133
#> 2 82 87 88 78 103 79 92 85 66 83
#> 3 132 147 155 154 147 143 138 131 144 147
#> spc.1786 spc.1790 spc.1794 spc.1798
#> 1 139 123 119 129
#> 2 75 87 92 90
#> 3 143 120 149 154To select one column, the $ operator is more convenient:
flu$c
#> [1] 0.05 0.10 0.15 0.20 0.25 0.30Class hyperSpec supports command line completion for the $ operator.
The extra data may also be set this way:
flu$n <- list(1:6, label = "sample no.")This function will append new columns, if necessary.
3.5 More on the [[]] and [[]]<-
3.5.1 Operators: Accessing Single Elements of the Spectra Matrix
Operator [[]] works mostly analogous to [].
In addition, however, these two functions also accept index matrices of size \(n × 2\).
In this case, a vector of values from the spectra matrix is returned.
indexmatrix <- matrix(c(1:3, 1:3), ncol = 2)
indexmatrix
#> [,1] [,2]
#> [1,] 1 1
#> [2,] 2 2
#> [3,] 3 3
faux_cell[[indexmatrix, wl.index = TRUE]]
#> [1] 132 73 142
diag(faux_cell[[1:3, , min ~ min + 2i]])
#> [1] 132 73 142Operator [[]]<- also accepts index matrices of size \(n × 2\).
3.6 Wavelengths
3.6.1 Converting Wavelengths to Indices and Vice Versa
Spectra in package hyperSpec always have discrete wavelength axes and are stored in a matrix with each column corresponding to one wavelength.
Two functions are provided to convert the respective column indices into wavelengths and vice versa: i2wl() and wl2i().
For i2wl() you should provide a vector of integers to serve as the indices. For wl2i() you can provide a vector of integers giving the wavelength range, or you can use a formula interface.
The basic syntax for the formula is start ~ end.
This yields a vector index_of_start : index_of_end.
The result of the formula conversion differs from the integer vector conversion in three ways:
- The colon operator for constructing vectors accepts only integer numbers, the tilde (for formulas) does not have this restriction.
- If the vector does not take into account the spectral resolution, one may get only every \(n\)th point or repetitions of the same index:
wl2i(flu, 405:410)
#> [1] 1 3 5 7 9 11
wl2i(flu, 405 ~ 410)
#> [1] 1 2 3 4 5 6 7 8 9 10 11
wl2i(faux_cell, 1000:1010)
#> [1] 100 101 101 101 101 102 102 102 102 103 103
wl2i(faux_cell, 1000 ~ 1010)
#> [1] 100 101 102 103- If the object’s wavelength axis is not ordered, the formula approach will give weird results.
In that (probably rare) case, use
wl_sort()first to obtain an object with an ordered wavelength axis.
Values start and end may contain the special variables min and max that
correspond to the lowest and highest wavelengths of the object:
wl2i(flu, min ~ 410)
#> [1] 1 2 3 4 5 6 7 8 9 10 11Often, specifications like “wavelength \(pm n\) data points” are needed. They can be given using complex numbers in the formula. The imaginary part is added to the index calculated from the wavelength in the real part:
wl2i(flu, 450 - 2i ~ 450 + 2i)
#> [1] 89 90 91 92 93
wl2i(flu, max - 2i ~ max)
#> [1] 179 180 181To specify several wavelength ranges, use a list containing the formulas and vectors1:
This mechanism also works for the wavelength arguments of [], [[]], and plot_spc().
3.6.2 Selecting Wavelength Ranges
Wavelength ranges can easily be selected using []’s third argument (Fig. 3.4).
plot(paracetamol[, , 2800 ~ 3200])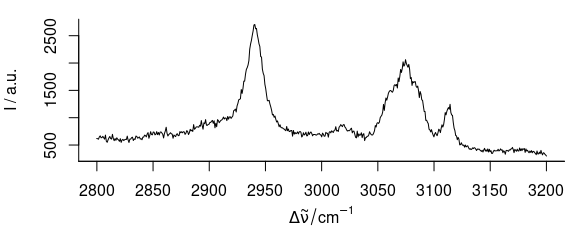
Figure 3.4: Spectra of paracetamol in range of 2800–3200 cm-1.
By default, the values given are treated as wavelengths.
If they are indices of the spectra matrix columns, use wl.index = TRUE:
plot(paracetamol[, , 2800:3200, wl.index = TRUE])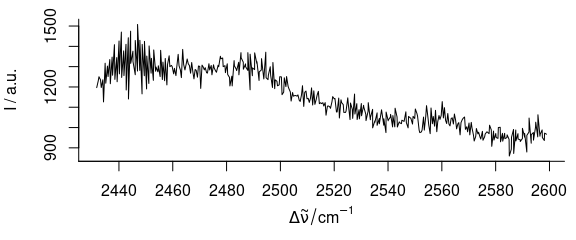
Figure 3.5: Spectra of paracetamol: from 2800th to 3200th point on wavelength axis.
Section 3.6.1 delves into the different possibilities of specifying wavelengths.
3.6.3 Deleting Wavelength Ranges
Deleting wavelength ranges may be accomplished using negative index vectors together with wl.index = TRUE.
plot(paracetamol[, , -(500:1000), wl.index = TRUE])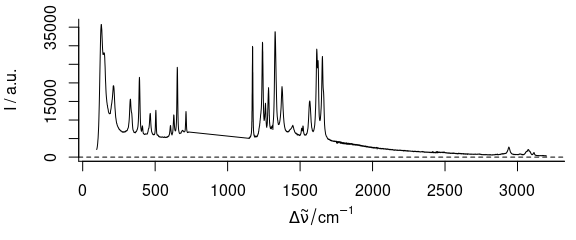
Figure 3.6: Spectra of paracetamol with 500th to 1000thwavelength axis point removed.
However, this mechanism works only if the proper indices are known.
If the range to be removed is instead specified in the units of the wavelength axis, it is easier to select the remainder of the spectrum instead. To delete the spectral range from 1750 to 2800 cm-1 of the paracetamol spectrum one can use:
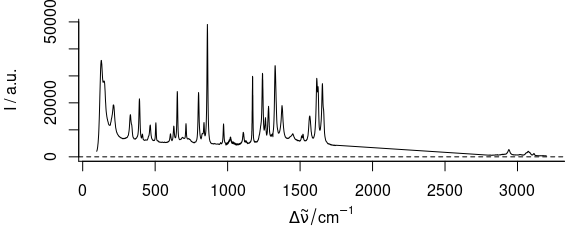
Figure 3.7: Spectra of paracetamol with range from 1750 to 2800 cm-1 removed.
It is possible to produce a plot of this data where the cut range is actually omitted and the wavelength axis is optionally cut in order to save space. For details see the plotting vignette.
3.6.4 Changing the Wavelength Axis
Sometimes wavelength axes need to be transformed, e.g., converting from wavelengths to frequencies.
In this case, retrieve the wavelength axis vector with wl(), convert each value of the resulting vector and assign the result with wl<-.
Also, the label of the wavelength axis may need to be adjusted.
As an example, convert the wavelength axis of laser to frequencies.
As the wavelengths are in nanometers, and the frequencies are easiest expressed in terahertz, an additional conversion factor of 1000 is needed:
laser
#> hyperSpec object
#> 84 spectra
#> 3 data columns
#> 36 data points / spectrum
wavelengths <- wl(laser)
frequencies <- 2.998e8 / wavelengths / 1000
wl(laser) <- frequencies
labels(laser, ".wavelength") <- "f / THz"
laser
#> hyperSpec object
#> 84 spectra
#> 3 data columns
#> 36 data points / spectrum
rm(laser)You can also accomplish these steps in a single line, e.g.,
wl(laser, "f / THz") <- frequenciesand
see ?wl<- for more information.
3.6.5 Ordering the Wavelength Axis
If the wavelength axis of an object needs reordering (e.g., after collapse()), wl_sort() can be used:
barb <- collapse(barbiturates[1:3])
wl(barb)
#> [1] 27.05 27.15 28.05 28.15 29.05 30.05 30.15 31.15 32.15 39.00 40.00 40.10 41.10
#> [14] 43.05 43.85 43.95 44.05 55.00 55.10 56.00 56.10 57.10 68.90 69.00 69.10 70.00
#> [27] 71.10 71.90 72.00 77.00 82.95 83.05 84.15 85.05 91.00 96.95 98.95 105.10 105.90
#> [40] 106.00 112.90 113.00 116.95 117.95 118.05 119.05 119.15 119.95 120.05 130.90 131.00 132.95
#> [53] 133.05 140.90 147.00 158.85 160.90
barb <- wl_sort(barb)
wl(barb)
#> [1] 27.05 27.15 28.05 28.15 29.05 30.05 30.15 31.15 32.15 39.00 40.00 40.10 41.10
#> [14] 43.05 43.85 43.95 44.05 55.00 55.10 56.00 56.10 57.10 68.90 69.00 69.10 70.00
#> [27] 71.10 71.90 72.00 77.00 82.95 83.05 84.15 85.05 91.00 96.95 98.95 105.10 105.90
#> [40] 106.00 112.90 113.00 116.95 117.95 118.05 119.05 119.15 119.95 120.05 130.90 131.00 132.95
#> [53] 133.05 140.90 147.00 158.85 160.90
3.7 Conversion to Long- and Wide-Format data.frames
The function as.data.frame() extracts the @data slot as a data.frame:
flu <- flu[, , 400 ~ 407] # make a small and handy version of the flu data set
as.data.frame(flu)
#> spc.405 spc.405.5 spc.406 spc.406.5 spc.407 filename c n .row
#> 1 27.150 32.345 33.379 34.419 36.531 rawdata/flu1.txt 0.05 1 1
#> 2 66.801 63.715 66.712 69.582 72.530 rawdata/flu2.txt 0.10 2 2
#> 3 93.144 103.068 106.194 110.186 113.249 rawdata/flu3.txt 0.15 3 3
#> 4 130.664 139.998 143.798 148.420 152.133 rawdata/flu4.txt 0.20 4 4
#> 5 167.267 171.898 177.471 184.625 189.752 rawdata/flu5.txt 0.25 5 5
#> 6 198.430 209.458 215.785 224.587 232.528 rawdata/flu6.txt 0.30 6 6
colnames(as.data.frame(flu))
#> [1] "spc" "filename" "c" "n" ".row"
as.data.frame(flu)$spc
#> 405 405.5 406 406.5 407
#> [1,] 27.150 32.345 33.379 34.419 36.531
#> [2,] 66.801 63.715 66.712 69.582 72.530
#> [3,] 93.144 103.068 106.194 110.186 113.249
#> [4,] 130.664 139.998 143.798 148.420 152.133
#> [5,] 167.267 171.898 177.471 184.625 189.752
#> [6,] 198.430 209.458 215.785 224.587 232.528Note that the spectra matrix is still a matrix inside column $spc.
Function as.data.frame() and the abbreviations $. and $.. retrieve the usual wide format data.frames:
flu$.
#> spc.405 spc.405.5 spc.406 spc.406.5 spc.407 filename c n
#> 1 27.150 32.345 33.379 34.419 36.531 rawdata/flu1.txt 0.05 1
#> 2 66.801 63.715 66.712 69.582 72.530 rawdata/flu2.txt 0.10 2
#> 3 93.144 103.068 106.194 110.186 113.249 rawdata/flu3.txt 0.15 3
#> 4 130.664 139.998 143.798 148.420 152.133 rawdata/flu4.txt 0.20 4
#> 5 167.267 171.898 177.471 184.625 189.752 rawdata/flu5.txt 0.25 5
#> 6 198.430 209.458 215.785 224.587 232.528 rawdata/flu6.txt 0.30 6
flu$..
#> filename c n
#> 1 rawdata/flu1.txt 0.05 1
#> 2 rawdata/flu2.txt 0.10 2
#> 3 rawdata/flu3.txt 0.15 3
#> 4 rawdata/flu4.txt 0.20 4
#> 5 rawdata/flu5.txt 0.25 5
#> 6 rawdata/flu6.txt 0.30 6If another subset of colums needs to be extracted, use [[]]:
flu[[, c("c", "spc")]]
#> c spc.405 spc.405.5 spc.406 spc.406.5 spc.407
#> 1 0.05 27.150 32.345 33.379 34.419 36.531
#> 2 0.10 66.801 63.715 66.712 69.582 72.530
#> 3 0.15 93.144 103.068 106.194 110.186 113.249
#> 4 0.20 130.664 139.998 143.798 148.420 152.133
#> 5 0.25 167.267 171.898 177.471 184.625 189.752
#> 6 0.30 198.430 209.458 215.785 224.587 232.528This can be combined with extracting certain spectra and wavelengths, see 3.8.
The transpose of a wide format data.frame can be obtained by as.t.df().
For further examples, see the discussion of package ggplot2 in the plotting vignette.
as.t.df(apply(flu, 2, mean_pm_sd))
#> .wavelength mean.minus.sd mean mean.plus.sd
#> spc.405 405.0 49.958 113.91 177.86
#> spc.405.5 405.5 53.396 120.08 186.77
#> spc.406 406.0 55.352 123.89 192.43
#> spc.406.5 406.5 57.310 128.64 199.96
#> spc.407 407.0 59.513 132.79 206.06Some functions need the data in an unstacked or long-format data.frame.
The function as.long.df() is the appropriate conversion function.
head(as.long.df(flu), 20)
#> .wavelength spc filename c n
#> 1 405.0 27.150 rawdata/flu1.txt 0.05 1
#> 2 405.0 66.801 rawdata/flu2.txt 0.10 2
#> 3 405.0 93.144 rawdata/flu3.txt 0.15 3
#> 4 405.0 130.664 rawdata/flu4.txt 0.20 4
#> 5 405.0 167.267 rawdata/flu5.txt 0.25 5
#> 6 405.0 198.430 rawdata/flu6.txt 0.30 6
#> 1.1 405.5 32.345 rawdata/flu1.txt 0.05 1
#> 2.1 405.5 63.715 rawdata/flu2.txt 0.10 2
#> 3.1 405.5 103.068 rawdata/flu3.txt 0.15 3
#> 4.1 405.5 139.998 rawdata/flu4.txt 0.20 4
#> 5.1 405.5 171.898 rawdata/flu5.txt 0.25 5
#> 6.1 405.5 209.458 rawdata/flu6.txt 0.30 6
#> 1.2 406.0 33.379 rawdata/flu1.txt 0.05 1
#> 2.2 406.0 66.712 rawdata/flu2.txt 0.10 2
#> 3.2 406.0 106.194 rawdata/flu3.txt 0.15 3
#> 4.2 406.0 143.798 rawdata/flu4.txt 0.20 4
#> 5.2 406.0 177.471 rawdata/flu5.txt 0.25 5
#> 6.2 406.0 215.785 rawdata/flu6.txt 0.30 6
#> 1.3 406.5 34.419 rawdata/flu1.txt 0.05 1
#> 2.3 406.5 69.582 rawdata/flu2.txt 0.10 23.8 Conversion to Matrix
To obtain the spectral data as a matrix, simply use the [[]] extractor:
flu[[]]
#> 405 405.5 406 406.5 407
#> [1,] 27.150 32.345 33.379 34.419 36.531
#> [2,] 66.801 63.715 66.712 69.582 72.530
#> [3,] 93.144 103.068 106.194 110.186 113.249
#> [4,] 130.664 139.998 143.798 148.420 152.133
#> [5,] 167.267 171.898 177.471 184.625 189.752
#> [6,] 198.430 209.458 215.785 224.587 232.528
class(flu[[]])
#> [1] "matrix" "array"Operator [[]] takes the same arguments as [], and can be used to extract a matrix containing parts of the spectra matrix:
flu[[1:3, , 406 ~ 407]]
#> 406 406.5 407
#> [1,] 33.379 34.419 36.531
#> [2,] 66.712 69.582 72.530
#> [3,] 106.194 110.186 113.249If wavelengths or indices for the columns to extract are given, a data.frame is returned instead of a matrix:
flu[[1:3, c("filename", "spc"), 406 ~ 407]]
#> filename spc.406 spc.406.5 spc.407
#> 1 rawdata/flu1.txt 33.379 34.419 36.531
#> 2 rawdata/flu2.txt 66.712 69.582 72.530
#> 3 rawdata/flu3.txt 106.194 110.186 113.2494 Creating a hyperSpec Object, Data Import and Export
Package hyperSpec comes with filters for a variety of file formats. These are discussed in detail in a separate fileio vignette. Here we just mention a couple of basic operations.
4.1 Creating a hyperSpec Object from Spectra Matrix and Wavelength Vector
If the data is in R’s workspace, a hyperSpec object is created by:
spc <- new("hyperSpec", spc = spectra.matrix, wavelength = wavelength.vector, data = extra.data)The most frequently needed arguments are:
spc |
The spectra matrix; may be a matrix or a data.frame. |
wavelength |
The wavelength axis as a numeric vector. |
data |
Any extra data as a data.frame.
It is possible to pass spc via this argument if the data frame has a column
$spc. |
label |
A list with the proper labels.
Do not forget the wavelength axis label in $.wavelength and the spectral
intensity axis label in $spc. |
More information about converting existing data into hyperSpec objects can be found in the fileio vignette.
4.2 Creating Random Spectra
If package mvtnorm[1,2] is available, multivariate normally distributed spectra can be generated from mean and covariance matrix using rmmvnorm() (Fig. 4.1).
Note that the package hyperSpec function’s name has an additional “m”: it already takes care of multiple groups.
Mean spectra and pooled covariance matrix can be calculated using cov_pooled():
pcov <- cov_pooled(faux_cell, faux_cell$region)
rnd <- rmmvnorm(rep(10, 3), mean = pcov$mean, sigma = pcov$COV)
Figure 4.1: Multivariate normally distributed random spectra generated with rmmvnorm().
5 Spectral Pre-Processing
5.1 Cutting the Spectral Range
Please refer to Table 3.1, Table 3.2 and Section 3.6 for information and examples of cutting the spectral range.
5.2 Shifting Spectra
Sometimes, spectra need to be aligned along the spectral axis.
In general, two options are available for shifting spectra along the wavelength axis.
- The wavelength axis can be shifted, while the intensities stay unaffected.
- The spectra are interpolated onto a new wavelength axis, while the nominal wavelengths stay.
The first method is very straightforward (Fig. 5.1):
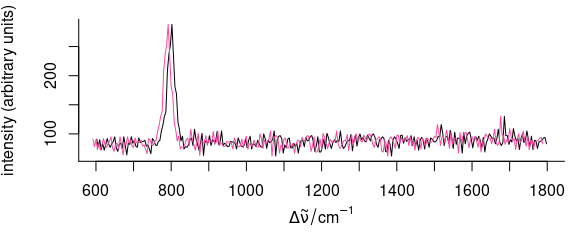
Figure 5.1: Shifting the spectra along the wavelength axis: changing the wavelength values.
However, the method cannot be used if each spectrum (or groups of spectra) are shifted individually.
In that case, interpolation is needed.
R offers many possibilities to interpolate (e.g., approx() for constant/linear approximation, spline() for spline interpolation, loess() can be used to obtain smoothed approximations, etc.).
The appropriate interpolation strategy will depend on the spectra, and package hyperSpec therefore leaves it up to the user to select a sensible interpolation function.
As an example, we will use natural splines to do the interpolation. It is convenient to set it up as a function:
interpolate <- function(spc, shift, wl) {
spline(wl + shift, spc, xout = wl, method = "natural")$y
}This function can now be applied to a set of spectra (Fig. 5.2):
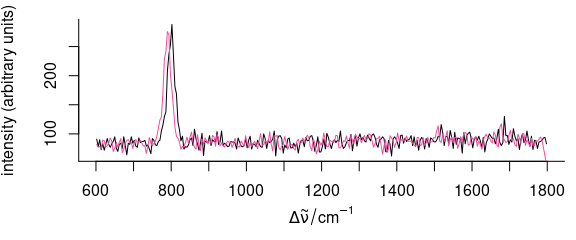
Figure 5.2: Shifting the spectra along the wavelength axis: interpolation.
Fig. 5.3 demonstrates the difference between simple shifting and interpolation.
tmp <- faux_cell[135, , 990 ~ 1010]
plot(tmp, lines.args = list(type = "b", pch = 19, cex = 0.5))
wl(tmp) <- wl(tmp) - 0.5
plot(tmp, lines.args = list(type = "b", pch = 19, cex = 0.5), add = TRUE, col = palette_colorblind[4])
tmp <- faux_cell[135]
tmp <- apply(tmp, 1, function(x, wl, shift) {
spline(wl + shift, x, xout = wl)$y
},
wl = wl(tmp), shift = -0.5
)
plot(tmp, lines.args = list(type = "b", pch = 19, cex = 0.5), add = TRUE, col = palette_colorblind[3])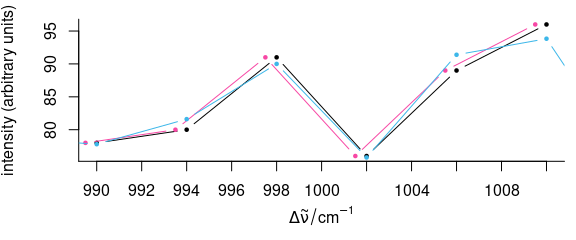
Figure 5.3: Shifting the spectra along the wavelength axis.
Detail view of the phenylalanine band: shifting by wl<- (red) does not affect the intensities, while the spectrum is slightly changed by interpolations (blue).
If different spectra need to be offset by a different shift, use a loop2.
shifts <- rnorm(nrow(faux_cell))
tmp <- faux_cell[[]]
for (i in seq_len(nrow(faux_cell))) {
tmp[i, ] <- interpolate(tmp[i, ], shifts[i], wl = wl(faux_cell))
}
faux_cell[[]] <- tmp5.2.1 Calculating the Shift
Often, the shift in the spectra is determined by aligning a particular signal. This strategy works best with spectrally oversampled data that allows accurate determination of the signal position.
For the faux_cell data, let’s use the maximum of the peak around 1200 cm-1.
As just the very maximum is too coarse, we’ll use the maximum of a square polynomial fitted to the maximum and its two neighbours.
# FIXME: this code currently does not work.
find_max <- function(y, x) {
pos <- which.max(y) + (-1:1)
X <- x[pos] - x[pos[2]]
Y <- y[pos] - y[pos[2]]
X <- cbind(1, X, X^2)
coef <- qr.solve(X, Y)
-coef[2] / coef[3] / 2 + x[pos[2]]
}
bandpos <- apply(faux_cell[[, , 1190 ~ 1210]], 1, find_max, wl(faux_cell[, , 1190 ~ 1210]))
refpos <- find_max(colMeans(faux_cell[[, , 1190 ~ 1210]]), wl(faux_cell[, , 1190 ~ 1210]))
shift1 <- refpos - bandposA second possibility is to optimize the shift. For this strategy, the spectra must be sufficiently similar, while low spectral resolution is compensated by using larger spectral windows.
faux_cell_tmp <- faux_cell - spc_fit_poly_below(faux_cell[, , min + 3i ~ max - 3i], faux_cell)
faux_cell_tmp <- sweep(faux_cell_tmp, 1, rowMeans(faux_cell[[]], na.rm = TRUE), "/")
targetfn <- function(shift, wl, spc, targetspc) {
error <- spline(wl + shift, spc, xout = wl)$y - targetspc
sum(error^2)
}
shift2 <- numeric(nrow(faux_cell))
tmp <- faux_cell[[]]
target <- colMeans(faux_cell[[]])
for (i in 1:nrow(faux_cell)) {
shift2[i] <- unlist(optimize(targetfn,
interval = c(-5, 5),
wl = faux_cell@wavelength,
spc = tmp[i, ], targetspc = target
)$minimum)
}Figure ?? shows that the second correction method works better for the faux_cell data.
This was expected, as the spectra are hardly or not oversampled, but are very similar to each other.
# FIXME: this code currently does not work.
df <- data.frame(
shift = c(shifts, shifts + shift1, shifts + shift2),
method = rep(c("original", "find maximum", "interpolation"),
each = nrow(faux_cell)
)
)
plot(histogram(~ shift | method,
data = df, breaks = do.breaks(range(df$shift), 25),
layout = c(3, 1)
))5.3 Removing Bad Data
5.3.1 Bad Spectra
Occasionally, one may want to remove spectra because of too low or too high signal.
E.g., for infrared spectra one may state that the absorbance maximum should be, say, between 0.1 and 1.
Class hyperSpec’s comparison operators return a logical matrix of the size of the spectra that is suitable for later indexing:
5.3.2 Removing Spectra Outside Mean \(\pm\) \(n\) Standard Deviations
mean_sd_filter <- function(x, n = 5) {
x <- x - mean(x)
s <- n * sd(x)
(x <= s) & (x > -s)
}
OK <- apply(faux_cell[[]], 2, mean_sd_filter, n = 4) # logical matrix
spc.OK <- faux_cell[apply(OK, 1, all)]
plot(faux_cell[!apply(OK, 1, all)])
i <- which(!OK, arr.ind = TRUE)
points(wl(faux_cell)[i[, 2]], faux_cell[[!OK]], pch = 19, col = palette_colorblind[4], cex = 0.5)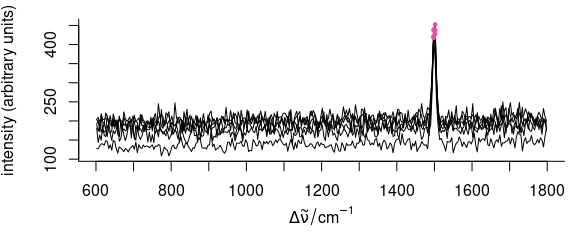
Figure 5.4: Filtering data: mean \(\pm\) sd filter.
5.3.3 Bad Data Points
Assume the data occasionally has a detector readout of 0:
spc <- faux_cell[1:3, , min ~ min + 15i]
spc[[cbind(1:3, sample(nwl(spc), 3)), wl.index = TRUE]] <- 0
spc[[]]
#> 602 606 610 614 618 622 626 630 634 638 642 646
#> [1,] 138.13 129.659 138.745 144.504 131.23 136.949 111.078 124.11 123.946 0.000 126.461 136.325
#> [2,] 0.00 73.371 99.227 83.301 54.00 74.184 68.029 79.00 73.236 98.166 82.623 73.986
#> [3,] 120.10 132.189 0.000 117.864 125.13 122.796 111.221 146.10 121.732 123.221 142.052 137.024
#> 650 654 658 662
#> [1,] 142.723 150.316 140.794 125.848
#> [2,] 72.874 69.232 90.187 83.842
#> [3,] 140.820 113.921 127.047 120.082We can set these points to NA, again using that the comparison returns a suitable logical matrix:
spc[[spc < 1e-4]] <- NA
spc[[]]
#> 602 606 610 614 618 622 626 630 634 638 642 646
#> [1,] 138.13 129.659 138.745 144.504 131.23 136.949 111.078 124.11 123.946 NA 126.461 136.325
#> [2,] NA 73.371 99.227 83.301 54.00 74.184 68.029 79.00 73.236 98.166 82.623 73.986
#> [3,] 120.10 132.189 NA 117.864 125.13 122.796 111.221 146.10 121.732 123.221 142.052 137.024
#> 650 654 658 662
#> [1,] 142.723 150.316 140.794 125.848
#> [2,] 72.874 69.232 90.187 83.842
#> [3,] 140.820 113.921 127.047 120.082Depending on the type of analysis, one may wish to replace the NAs by interpolating the neighbour values.
Package hyperSpec provides three functions that can interpolate the NAs: spc_na_approx(), spc_loess(), and spc_bin() with na.rm = TRUE (the latter two are discussed below).
spc.corrected <- spc_na_approx(spc)
spc.corrected[[]]
#> 602 606 610 614 618 622 626 630 634 638 642 646
#> [1,] 138.127 129.659 138.745 144.504 131.23 136.949 111.078 124.11 123.946 125.204 126.461 136.325
#> [2,] 73.371 73.371 99.227 83.301 54.00 74.184 68.029 79.00 73.236 98.166 82.623 73.986
#> [3,] 120.105 132.189 125.026 117.864 125.13 122.796 111.221 146.10 121.732 123.221 142.052 137.024
#> 650 654 658 662
#> [1,] 142.723 150.316 140.794 125.848
#> [2,] 72.874 69.232 90.187 83.842
#> [3,] 140.820 113.921 127.047 120.082
spc[[is.na(spc)]] <- 0
plot(spc)
spc[[spc < 1e-4]] <- NA
plot(spc_na_approx(spc),
add = TRUE, col = palette_colorblind[4],
lines.args = list(type = "b", pch = 19, cex = 0.5)
)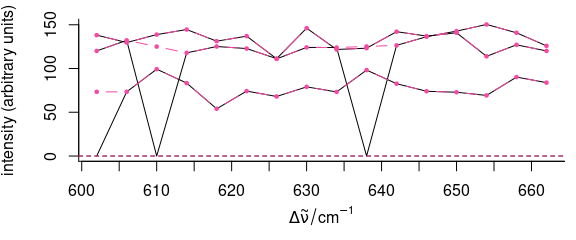
Figure 5.5: Filtering data: remove bad points.
5.4 Smoothing Interpolation
Spectra acquired by grating instruments are frequently interpolated onto a new wavelength axis, e.g. because the unequal data point spacing should be removed.
Also, the spectra can be smoothed: reducing the spectral resolution allows for an increase in the signal-to-noise ratio.
For chemometric data analysis reducing the number of data points per spectrum may be crucial as it reduces the dimensionality of the data.
Package hyperSpec provides two functions to do so: spc_bin() and spc_loess().
Function spc_bin() bins the spectral axis by averaging every by data points.
plot(paracetamol, wl.range = c(300 ~ 1800, 2800 ~ max), xoffset = 850)
#> Loading required namespace: plotrix
p <- spc_loess(paracetamol, c(seq(300, 1800, 2), seq(2850, 3150, 2)))
plot(p, wl.range = c(300 ~ 1800, 2800 ~ max), xoffset = 850, col = palette_colorblind[4], add = TRUE)
b <- spc_bin(paracetamol, 4)
plot(b,
wl.range = c(300 ~ 1800, 2800 ~ max), xoffset = 850,
lines.args = list(pch = 20, cex = .3, type = "p"), col = palette_colorblind[3], add = TRUE
)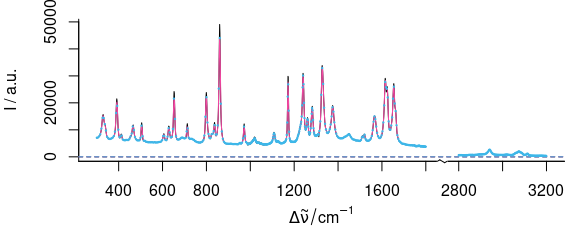
Figure 5.6: Smoothing interpolation by spc_loess() with new data point spacing of 2 cm-1 (red) and spc_bin() (blue).
plot(paracetamol[, , 1600 ~ 1670])
plot(p[, , 1600 ~ 1670], col = palette_colorblind[4], add = TRUE)
plot(b[, , 1600 ~ 1670], col = palette_colorblind[3], add = TRUE)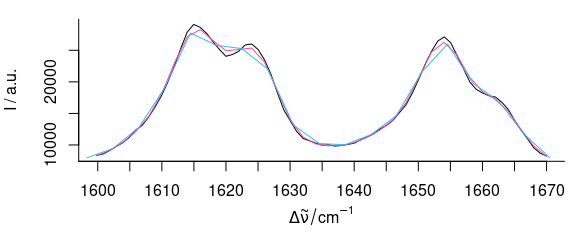
Figure 5.7: The magnification of Fig. 5.6 shows how interpolation may cause a loss in signal height.
Function spc_loess() applies R’s loess() function for spectral interpolation.
Figures 5.6 and 5.7 show the result of interpolating from 300 to 1800 and 2850 to 3150 cm-1 with 2 cm-1 data point distance.
This corresponds to a spectral resolution of about 4 cm-1, and the decrease in spectral resolution can be seen at the sharp bands where the maxima are not reached (due to the fact that the interpolation wavelength axis does not necessarily hit the maxima.
The original spectrum had 4064 data points with unequal data point spacing (between 0 and 1.4 cm-1).
The interpolated spectrum has 902 data points.
5.5 Background Correction
To subtract a background spectrum of each of the spectra in an object, use:
sweep(spectra, 2, background.spectrum, "-")5.6 Offset Correction
Calculate the offsets and adjust the spectra accordingly:
If the offset is calculated by a function, as here with the min(), hyperSpec’s sweep() method offers a shortcut: sweep()’s STATS argument may be the function instead of a numeric vector:
faux_cell_offset_corrected <- sweep(faux_cell, 1, min, "-")5.7 Baseline Correction
Package hyperSpec comes with two functions to fit polynomial baselines.
Functions spc_fit_poly_below(), spc_fit_poly() fit a polynomial baseline of the given order.
A least-squares fit is done so that the function may be used on rather noisy spectra.
However, the user must supply an object that is cut appropriately.
Particularly, the supplied wavelength ranges are not weighted.
The function spc_fit_poly_below() tries to find appropriate support points for the baseline iteratively.
Both functions return a hyperSpec object containing the fitted baselines.
They need to be subtracted afterward:
bl <- spc_fit_poly_below(faux_cell)
faux_cell_tmp <- faux_cell - blFor details, see baseline vignette.
Package package baseline[3,4] offers many more functions for baseline correction.
The baseline() function works on the spectra matrix, which is extracted by [[]].
The result is a baseline object, but can easily be re-imported into the hyperSpec object:
corrected <- hyperSpec::faux_cell[1] # start with the unchanged data set
library("baseline")
#>
#> Attaching package: 'baseline'
#> The following object is masked from 'package:pls':
#>
#> mvrValstats
#> The following object is masked from 'package:stats':
#>
#> getCall
bl <- baseline(corrected[[]], method = "modpolyfit", degree = 4)
corrected[[]] <- getCorrected(bl)Fig. 5.8 and 5.9 show raw data and the result for the first spectrum of faux_cell.
baseline <- corrected
baseline[[]] <- getBaseline(bl)
plot(hyperSpec::faux_cell[1], plot.args = list(ylim = range(hyperSpec::faux_cell[1], 0)))
plot(baseline, add = TRUE, col = palette_colorblind[4])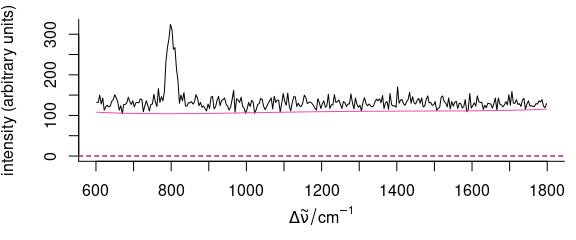
Figure 5.8: The first spectrum of faux_cell (raw data) with baseline.
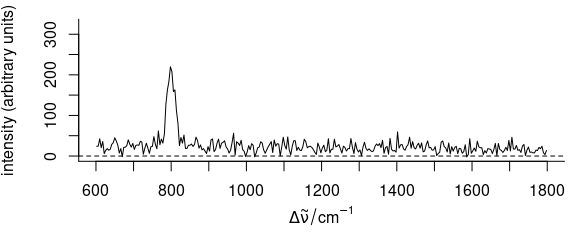
Figure 5.9: Baseline correction using the baseline package:
the first spectrum of faux_cell after baseline correction with method “odpolyfi”.
rm(bl, faux_cell)5.8 Intensity Calibration
5.8.1 Correcting by a Constant, e. g. Readout Bias
CCD cameras often operate with a bias, causing a constant value for each pixel.
Such a constant can be immediately subtracted: spectra - constant.
5.8.2 Correcting Wavelength Dependence
For each of the wavelengths, the same correction needs to be applied to all spectra.
5.9 Normalization
Again, sweep() is the function of choice.
E.g., for area normalization, use:
faux_cell_tmp <- sweep(faux_cell, 1, mean, "/")(Using the mean instead of the sum results in conveniently scaled spectra with intensities around 1.)
If the calculation of the normalization factors is more elaborate, use a two step procedure:
Calculate appropriate normalization factors You may calculate the factors using only a certain wavelength range, thereby normalizing on a particular band or peak.
-
Again, sweep the factor off the spectra:
normalized <- sweep(spectra, 1, factors, "*")
factors <- 1 / apply(faux_cell[, , 1600 ~ 1700], 1, mean)
faux_cell_tmp <- sweep(faux_cell, 1, factors, "*")For the special case of area normalization using the mean spectra, the factors can be more conveniently calculated by
factors <- 1 / rowMeans(faux_cell[, , 1600 ~ 1700])and instead of sweep the arithmetic operators (here *) can be used directly with the normalization factor:
faux_cell_tmp <- faux_cell * factorsPut together, this results in:
faux_cell_tmp <- faux_cell / rowMeans(faux_cell[, , 1600 ~ 1700])For minimum-maximum-normalization, first do an offset- or baseline correction, then normalize using max().
5.10 Centering and Variance Scaling the Spectra
Centering means that the mean spectrum is subtracted from each of the spectra. Many data analysis techniques, like principal component analysis, partial least squares, etc., work much better on centered data. From a spectroscopic point of view, it depends on the particular data set whether centering does make sense or not.
Variance scaling is often used in multivariate analysis to adjust the influence and scaling of the variates (that are typically different physical values). However, spectra already have the same scale of the same physical value. Thus one has to trade off the the expected numeric benefit with the fact that for wavelengths with low signal, the noise level will greatly increase when using variance scaling. Scaling usually makes sense only for centered data.
Both tasks are carried out by the same method in R, scale(), which will by default both mean center and variance scale the spectra matrix.
To center the flu data set, use:
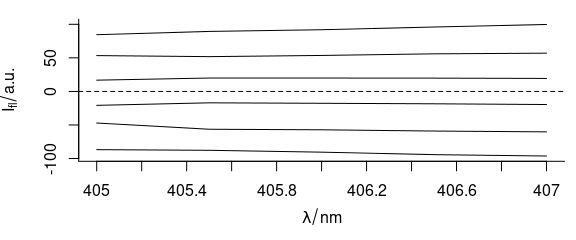
Figure 5.10: Mean-centered spectra of flu.
On the other hand, the faux_cell data set consists of Raman spectra, so the spectroscopic interpretation of centering is getting rid of the average chemical composition of the sample.
But what is the meaning of the “Average spectrum” of an inhomogeneous sample?
In this case, it may be better to subtract the minimum spectrum (which will hopefully have almost the same benefit on the data analysis) as it is the spectrum of that chemical composition that is underlying the whole sample.
One more point to consider is that the actual minimum spectrum will pick up (negative) noise. To avoid that, using, e.g., the 5th percentile spectrum is more suitable:
faux_cell_tmp <- scale(faux_cell, center = quantile(faux_cell, 0.05), scale = FALSE)
plot(faux_cell_tmp, "spcprctl5")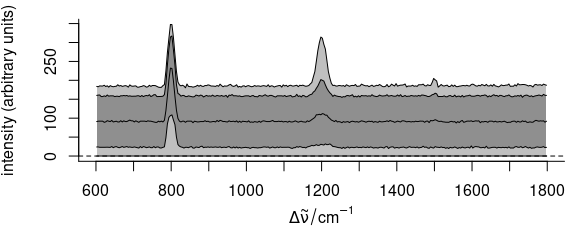
Figure 5.11: The summary spectra of faux_cell with 5th percentile subtracted.
See section the appendices for some tips to speed up these calculations.
5.11 Multiplicative Scatter Correction
Multiplicative scatter correction (MSC) can be done using msc() from package pls [5].
It operates on the spectra matrix:
5.12 Spectral Arithmetic
Basic mathematical functions are defined for hyperSpec objects.
You may convert spectra to their log:
absorbance.spectra <- -log10(transmission.spectra)In this case, do not forget to adapt the label:
labels(absorbance.spectra)$spc <- "A"Be careful: R’s log() function calculates the natural logarithm if no base is given.
The basic arithmetic operators work element-wise in R.
Thus they all need either a scalar or a matrix (or hyperSpec object) of the correct size.
Matrix multiplication is done by %*%, again each of the operands may be a matrix or a hyperSpec object and must have the correct dimensions.
There are many more mathematical functions that understand a hyperSpec object.
See ?Arith for more details.
6 Data Analysis
6.1 Principal Component Analysis with prcomp()
6.1.1 Carrying out PCA
The $. notation is handy, if a data analysis function expects a data.frame, as prcomp() does.
The column names can then be used in the formula:
pca <- prcomp(~spc, data = faux_cell$., center = FALSE)However, many modeling functions call as.data.frame on their data argument, including prcomp().
In that case, the conversion is done automatically so the following gives the same result as above:
pca <- prcomp(~spc, data = faux_cell, center = FALSE)6.1.2 Plotting the Results of PCA
The results of such a decomposition can be put back into hyperSpec objects.
This conveniently allows one to use the regular hyperSpec plotting functions, e.g., the loading-like spectra, or score maps, see Figures 6.1 and 6.2.
scores <- decomposition(faux_cell, pca$x,
label.wavelength = "PC",
label.spc = "score / a.u."
)
scores
#> hyperSpec object
#> 875 spectra
#> 4 data columns
#> 300 data points / spectrumThe loadings can be similarly re-imported:
loadings <- decomposition(faux_cell, t(pca$rotation),
scores = FALSE,
label.spc = "loading I / a.u."
)
loadings
#> hyperSpec object
#> 300 spectra
#> 1 data columns
#> 300 data points / spectrumThere is, however, one important difference. The loadings are thought of as values computed from all spectra together. Thus no meaningful extra data can be assigned for the loadings object (at least not if the column consists of different values). Therefore, the loadings object lost all extra data (see above).
Argument retain.columns triggers whether columns that contain different values should be dropped.
If it is set to TRUE, the columns are retained, but contain NAs:
loadings <- decomposition(faux_cell, t(pca$rotation),
scores = FALSE,
retain.columns = TRUE, label.spc = "loading I / a.u."
)
loadings[1]$..
#> x y region
#> PC1 NA NA <NA>If an extra data column does contain only one unique value, it is retained regardless:
faux_cell$measurement <- 1
loadings <- decomposition(faux_cell, t(pca$rotation),
scores = FALSE,
label.spc = "loading I / a.u."
)
loadings[1]$..
#> measurement
#> PC1 1
plot(loadings[1:3], stacked = TRUE)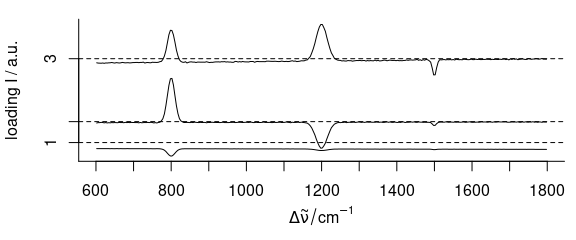
Figure 6.1: The first three loadings.
div_palette <- colorRampPalette(c(
"#00008B", "#351C96", "#5235A2", "#6A4CAE", "#8164BA", "#967CC5",
"#AC95D1", "#C1AFDC", "#D5C9E8", "#E0E3E3", "#F8F8B0", "#F7E6C2",
"#EFCFC6", "#E6B7AB", "#DCA091", "#D08977", "#C4725E", "#B75B46",
"#A9432F", "#9A2919", "#8B0000"
), space = "Lab")
plot_map(scores[, , 3], col.regions = diverging_hcl(20, palette = "Blue-Red2"))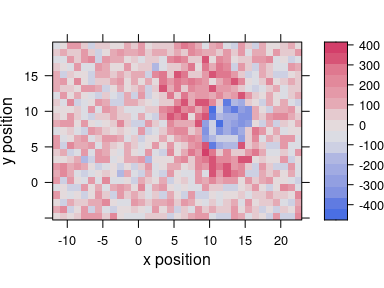
Figure 6.2: The third score map.
6.1.3 PCA as Noise Filter
Principal component analysis is sometimes used as a noise-filtering technique. The idea is that the relevant differences are captured in the first few components while the higher components contain noise only. Thus the spectra are reconstructed using only the first \(p\) components.
This reconstruction is a matrix multiplication:
\[\begin{equation} \mathrm{spectra}^{\mathrm{~nrow} ~×~ \mathrm{nwl}} = \mathrm{scores}^{\mathrm{~nrow} ~×~ p} \;\cdot\; \mathrm{loadings}^{~p ~×~ \mathrm{nwl}} \tag{6.1} \end{equation}\]
Note that this corresponds to a model based on the Beer-Lambert law: \[\begin{equation} A_n (\lambda) = c_{n,i} \epsilon (i, \lambda) + \mathrm{error} \tag{6.2} \end{equation}\] The matrix formulation puts the \(n\) spectra into the rows of \(A\) and \(c\), while the \(i\) pure components appear in the columns of \(c\) and rows of the absorbance coefficients \(\epsilon\).
For an ideal data set (constituents varying independently, sufficient signal-to-noise ratio) one would expect the principal component analysis to extract something like the concentrations and pure component spectra.
If we decide that only the first 10 components actually carry spectroscopic information, we can reconstruct spectra with a better signal-to-noise ratio:
smoothed <- scores[, , 1:10] %*% loadings[1:10]Keep in mind, though, that we cannot be sure how much useful information was discarded with the higher components. This kind of noise reduction may influence further modeling of the data. Mathematically speaking, the rank of the new 875 \(×\) 300 spectra matrix is only 10.
6.2 Hierarchical Cluster Analysis
Some R functions expect their input data in a matrix, including those related to HCA.
Conveniently, the [[]] extraction function returns a matrix.
dist <- dist_pearson(faux_cell[[]])Again, many such functions coerce the data to a matrix automatically, so the hyperSpec object can be handed over without the explicit conversion above (as we saw for PCA):
dist <- dist_pearson(faux_cell)
dendrogram <- hclust(dist, method = "ward.D")
plot(dendrogram, labels = FALSE, hang = -1)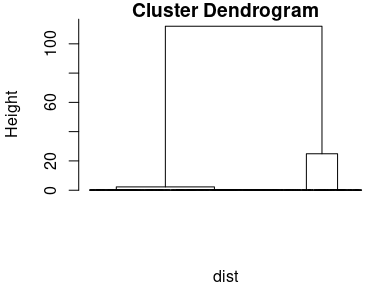
Figure 6.3: The results of the cluster analysis: the dendrogram.
In order to plot a cluster map, the cluster membership needs to be calculated from the dendrogram.
First, cut the dendrogram so that three clusters result:
As the cluster membership was stored as a factor, the levels can be meaningful names, which are displayed in the color legend.
Then the result may be plotted (Figure 6.4):
plot_map(faux_cell, region ~ x * y, col.regions = cluster.cols)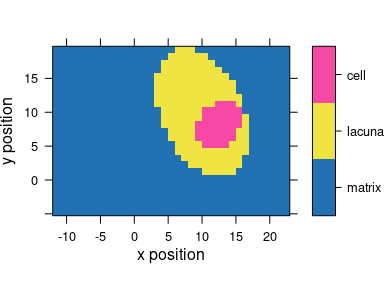
Figure 6.4: The results of the cluster analysis: the map of the 3 clusters.
6.3 Calculating Group-Wise Sum Characteristics, e. g. Cluster Mean Spectra
Function aggregate() applies the function given in FUN to each of the groups of spectra specified in by.
So we may plot the cluster mean spectra:
means <- aggregate(faux_cell, by = faux_cell$region, mean_pm_sd)
plot(means, col = cluster.cols, stacked = ".aggregate", fill = ".aggregate")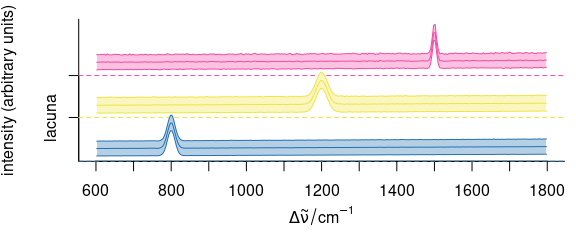
Figure 6.5: The results of the cluster analysis: the mean spectra.
7 Combining and Splitting hyperSpec Objects
7.1 Binding Objects Together
Class hyperspec objects can be bound together, either by columns (cbind()) to append a new spectral range or by row (rbind()) to append new spectra:
dim(flu)
#> nrow ncol nwl
#> 6 4 5There is also a more general function, bind(), taking the direction ("r" or "c") as the first argument followed by the objects to bind either in separate arguments or in a list.
As usual for rbind() and cbind(), the objects that should be bound together must have
the same number of columns (for rbind()) and the same number of rows (for cbind()),
respectively.
For binding row-wise (rbind()), collapse() is more flexible and faster as well.
7.2 Binding Objects that Do not Share the Same Extra Data and/or Wavelength Axis
Function collapse() combines objects that should be bound together by row, but they do not share the columns and/or spectral range.
The resulting object has all columns from all input objects and all wavelengths from the input objects.
If an input object does not have a particular column or wavelength, its value in the resulting object is NA.
The barbiturates data is a list of 5 hyperSpec objects, each containing one mass spectrum.
The spectra have between 22 and 37 data points each.
barb <- collapse(barbiturates)
wl(barb)[1:25]
#> [1] 27.05 27.15 28.05 28.15 29.05 30.05 30.15 31.15 32.15 38.90 39.00 40.00 40.10 41.10 43.05 43.85
#> [17] 43.95 44.05 55.00 55.10 56.00 56.10 57.00 57.10 68.90The resulting object does not have an ordered wavelength axis. This can be obtained in the second step:
barb <- wl_sort(barb)
barb[[1:3, , min ~ min + 10i]]
#> 27.05 27.15 28.05 28.15 29.05 30.05 30.15 31.15 32.15 38.9 39
#> [1,] 562 NA NA 11511 6146 1253 NA 9851 10751 NA 756
#> [2,] NA 618 10151 NA 5040 NA 910 8200 10989 NA 770
#> [3,] 638 NA NA 10722 5253 966 NA 8709 10899 NA NA7.3 Binding Objects that Do not Share the Same Spectra
The function merge() adds a new spectral range (like cbind()) but works also if spectra are missing in one of the objects.
The arguments by, by.x, and by.y specify which columns should be used to decide which spectra are the same.
The arguments all, all.x, and all.y determine whether spectra should be kept for the result set if they appear in only one of the objects.
For details, see also the help on the base function merge().
As an example, let’s construct a version of the faux_cell data like being taken as two maps with different spectral ranges.
In each data set, some spectra are missing.
As all extra data columns are the same, no special declarations are needed for merging the data:
By default, the result consists of only those spectra, where both spectral ranges were available.
To keep all spectra replacing missing parts by NA (see Fig. 7.1, 7.2):
faux_cell_merged <- merge(faux_cell_low, faux_cell_high, all = TRUE)
nrow(faux_cell_merged)
#> [1] 841
matcols <- sequential_hcl(20, palette = "viridis")
levelplot(spc ~ x * y | as.factor(paste(.wavelength, " 1/cm")),
faux_cell_merged[, , c(1000, 1650)],
aspect = "iso", col.regions = matcols
)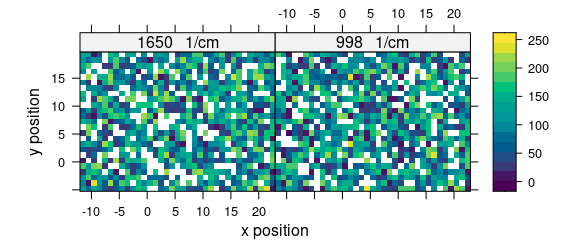
Figure 7.1: For both spectral ranges some spectra are missing.
plot(faux_cell_merged[1:100], "mat", col = matcols)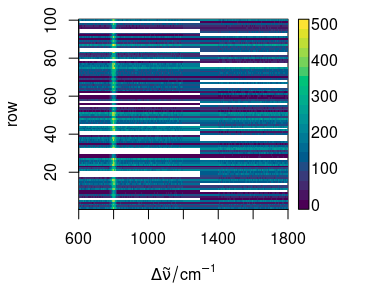
Figure 7.2: The missing parts of the spectra are filled with NA.
merged <- merge(faux_cell[1:7, , 610 ~ 620], faux_cell[5:10, , 615 ~ 625], all = TRUE)
merged$.
#> x y region measurement .nx .ny spc.1 spc.2 spc.3 spc.4 spc.5 spc.6 spc.7
#> 1 -11.55 -4.77 matrix 1 1 NA 150 128 143 NA NA NA NA
#> 2 -10.55 -4.77 matrix 1 2 NA 99 84 54 NA NA NA NA
#> 3 -9.55 -4.77 matrix 1 3 NA 142 118 125 NA NA NA NA
#> 4 -8.55 -4.77 matrix 1 4 NA 83 108 126 NA NA NA NA
#> 5 -7.55 -4.77 matrix 1 5 1 27 19 23 19 23 27 31
#> 6 -6.55 -4.77 matrix 1 6 2 14 18 12 18 12 20 17
#> 7 -5.55 -4.77 matrix 1 7 3 22 24 23 24 23 28 31
#> 8 -4.55 -4.77 matrix 1 NA 4 NA NA NA 74 78 86 81
#> 9 -3.55 -4.77 matrix 1 NA 5 NA NA NA 0 0 0 1
#> 10 -2.55 -4.77 matrix 1 NA 6 NA NA NA 122 129 135 119If the spectra overlap, the result will have both data points. In the example here one could easily delete duplicate wavelengths. For real data, however, the duplicated wavelength will hardly ever contain the same values. The appropriate method to deal with this situation depends on the data at hand, but it will usually be some kind of spectral interpolation.
One possibility is removing duplicated wavelengths by using the mean intensity.
This can conveniently be done by using approx() using method = "constant".
For duplicated wavelengths, the intensities will be combined by the tie function.
This already defaults to the mean, but we need na.rm = TRUE.
Thus, the function to calculate the new spectral intensities is
approxfun <- function(y, wl, new.wl) {
approx(wl, y, new.wl,
method = "constant",
ties = function(x) mean(x, na.rm = TRUE)
)$y
}which can be applied to the spectra:
merged <- apply(merged, 1, approxfun,
wl = wl(merged),
new.wl = unique(wl(merged)),
new.wavelength = "new.wl"
)
merged$.
#> x y region measurement .nx .ny spc.1 spc.2 spc.3 spc.4 spc.5
#> 1 -11.55 -4.77 matrix 1 1 NA 150 128 143 NA NA
#> 2 -10.55 -4.77 matrix 1 2 NA 99 84 54 NA NA
#> 3 -9.55 -4.77 matrix 1 3 NA 142 118 125 NA NA
#> 4 -8.55 -4.77 matrix 1 4 NA 83 108 126 NA NA
#> 5 -7.55 -4.77 matrix 1 5 1 27 19 23 27 31
#> 6 -6.55 -4.77 matrix 1 6 2 14 18 12 20 17
#> 7 -5.55 -4.77 matrix 1 7 3 22 24 23 28 31
#> 8 -4.55 -4.77 matrix 1 NA 4 NA 74 78 86 81
#> 9 -3.55 -4.77 matrix 1 NA 5 NA 0 0 0 1
#> 10 -2.55 -4.77 matrix 1 NA 6 NA 122 129 135 1197.4 Merging Extra Data to Objects That Do Not (Necessarily) Share the Same Spectra
Assume we obtained duplicate reference measurements for some of the concentrations in flu:
flu.ref <- data.frame(
filename = rep(flu$filename[1:2], each = 2),
cref = rep(flu$c[1:2], each = 2) + rnorm(4, sd = 0.01)
)
flu.ref
#> filename cref
#> 1 rawdata/flu1.txt 0.044329
#> 2 rawdata/flu1.txt 0.044622
#> 3 rawdata/flu2.txt 0.087589
#> 4 rawdata/flu2.txt 0.119215This information can be merged into the extra data of flu by:
flu.merged <- merge(flu, flu.ref)
flu.merged$..
#> filename c n cref
#> 1 rawdata/flu1.txt 0.05 1 0.044329
#> 2 rawdata/flu1.txt 0.05 1 0.044622
#> 3 rawdata/flu2.txt 0.10 2 0.087589
#> 4 rawdata/flu2.txt 0.10 2 0.119215The usual rules for merge() apply.
E.g. to preserve all spectra of flu, use all.x = TRUE:
flu.merged <- merge(flu, flu.ref, all.x = TRUE)
flu.merged$..
#> filename c n cref
#> 1 rawdata/flu1.txt 0.05 1 0.044329
#> 2 rawdata/flu1.txt 0.05 1 0.044622
#> 3 rawdata/flu2.txt 0.10 2 0.087589
#> 4 rawdata/flu2.txt 0.10 2 0.119215
#> 5 rawdata/flu3.txt 0.15 3 NA
#> 6 rawdata/flu4.txt 0.20 4 NA
#> 7 rawdata/flu5.txt 0.25 5 NA
#> 8 rawdata/flu6.txt 0.30 6 NAThe class of the first object (x) determines the resulting class:
7.5 Splitting an Object, and Binding a List of hyperSpec Objects
A hyperSpec object may also be split into a list of hyperSpec objects:
clusters <- split(faux_cell, faux_cell$region)
clusters
#> $matrix
#> hyperSpec object
#> 682 spectra
#> 5 data columns
#> 300 data points / spectrum
#>
#> $lacuna
#> hyperSpec object
#> 155 spectra
#> 5 data columns
#> 300 data points / spectrum
#>
#> $cell
#> hyperSpec object
#> 38 spectra
#> 5 data columns
#> 300 data points / spectrumSplitting can be reversed by rbind() (see section 7.1).
Another, similar way to combine a number of hyperSpec objects with different wavelength axes or extra data columns is collapse() (see section 7.2).
Both rbind() and collapse() take care that factor levels are expanded as necessary:
lacunae <- droplevels(faux_cell[faux_cell$region == "matrix" & !is.na(faux_cell$region)])
summary(lacunae$region)
#> matrix
#> 682
cells <- droplevels(faux_cell[faux_cell$region == "cell" & !is.na(faux_cell$region)])
summary(cells$region)
#> cell
#> 38
7.6 Factor Columns in hyperSpec Objects: Dropping Factor Levels That Are Not Needed
For subsections of hyperSpec objects that do not contain all levels of a factor column, droplevels() drops the “unpopulated” levels:
tmp <- faux_cell[1:50]
table(tmp$region)
#>
#> matrix lacuna cell
#> 50 0 0
tmp <- droplevels(tmp)
table(tmp$region)
#>
#> matrix
#> 508 Plotting
Package hyperSpec offers a variety of possibilities to plot spectra, spectral maps, the spectra matrix, time series, depth profiles, etc. See the plotting vignette.
9 Appendices
Package Options
| name | default value (range) | description | used by |
|---|---|---|---|
debuglevel |
0 (1L, 2L) |
Amount of debugging information produced |
identify_spc(), map.identify(), spc_rubberband(),various file import functions. |
gc |
FALSE |
Triggers frequent calling of gc()
|
read.ENVI(), new("hyperSpec")
|
tolerance |
sqrt(.Machine$.double.eps) |
Tolerance for numerical comparisons | File import functions (removing empty spectra), normalize_01()
|
wl.tolerance |
sqrt(.Machine$.double.eps) |
Tolerance for comparisons of the wavelength axis |
rbind(), rbind2(), bind("r", ...), all.equal(), collapse()
|
file.remove.emptyspc |
TRUE |
Automatic removing of empty spectra | File import functions, see fileio |
file.keep.name |
TRUE |
Automatic recording of file name in column filename
|
File import functions, see fileio |
plot.spc.nmax |
25 |
Number of spectra to be plotted by default | plot_spc() |
ggplot.spc.nmax |
10 |
qplotspc() |
Speed and Memory Considerations
While most of the package hyperSpec’s functions work at a decent speed for interactive sessions (of course depending on the size of the object), iterated (repeated) calculations for bootstrapping or iterated cross-validation may ask for special speed considerations.
As an example, consider the code for shifting the spectra:
tmp <- faux_cell[1:50]
shifts <- rnorm(nrow(tmp))
system.time({
for (i in seq_len(nrow(tmp))) {
tmp[[i]] <- interpolate(tmp[[i]], shifts[i], wl = wl(tmp))
}
})
#> user system elapsed
#> 0.037 0.000 0.037Calculations that involve a lot of subsetting (i.e., extracting or changing the spectra matrix or extra data) can be sped up considerably if the required parts of the hyperSpec object are extracted beforehand.
This is somewhat similar to model fitting in R in general: many model fitting functions in R are much faster if the formula interface is avoided and the appropriate data.frames or matrices are handed over directly.
tmp <- faux_cell[1:50]
system.time({
tmp.matrix <- tmp[[]]
wl <- wl(tmp)
for (i in seq_len(nrow(tmp))) {
tmp.matrix[i, ] <- interpolate(tmp.matrix[i, ], shifts[i], wl = wl)
}
tmp[[]] <- tmp.matrix
})
#> user system elapsed
#> 0.016 0.000 0.0179.1 Additional Packages
Package matrixStats[6] implements fast functions to calculate summary statistics for each row or each column of a matrix.
9.2 Memory Usage
In general, it is not recommended to work with variables that are more than approximately a third of the available RAM in size. Particularly the import of raw spectroscopic data can consume large amounts of memory. At certain points, package hyperSpec provides switches that allow working with data sets that are actually close to this memory limit.
The initialization method new("hyperSpec", ...) takes particular care to avoid unnecessary copies of the spectra matrix.
In addition, frequent calls to gc() can be requested by hy_set_option(gc = TRUE).
The same behaviour is triggered in read.ENVI() and its derivatives (read.ENVI.Manufacturer()).
The memory consumption of read.txt.Renishaw() can be lowered by importing the data in chunks (argument nlines).
Session Info
sessioninfo::session_info("hyperSpec")
#> ─ Session info ───────────────────────────────────────────────────────────────────────────────────
#> setting value
#> version R version 4.4.0 (2024-04-24)
#> os Ubuntu 22.04.4 LTS
#> system x86_64, linux-gnu
#> ui X11
#> language en
#> collate C.UTF-8
#> ctype C.UTF-8
#> tz UTC
#> date 2024-05-27
#> pandoc 3.1.11 @ /opt/hostedtoolcache/pandoc/3.1.11/x64/ (via rmarkdown)
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────────────────────────
#> ! package * version date (UTC) lib source
#> brio 1.1.5 2024-04-24 [2] RSPM
#> callr 3.7.6 2024-03-25 [2] RSPM
#> cli 3.6.2 2023-12-11 [2] RSPM
#> colorspace * 2.1-0 2023-01-23 [2] RSPM
#> crayon 1.5.2 2022-09-29 [2] RSPM
#> deldir 2.0-4 2024-02-28 [2] RSPM
#> desc 1.4.3 2023-12-10 [2] RSPM
#> diffobj 0.3.5 2021-10-05 [2] RSPM
#> digest 0.6.35 2024-03-11 [2] RSPM
#> dplyr 1.1.4 2023-11-17 [2] RSPM
#> evaluate 0.23 2023-11-01 [2] RSPM
#> fansi 1.0.6 2023-12-08 [2] RSPM
#> farver 2.1.2 2024-05-13 [2] RSPM
#> fs 1.6.4 2024-04-25 [2] RSPM
#> generics 0.1.3 2022-07-05 [2] RSPM
#> ggplot2 * 3.5.1 2024-04-23 [2] RSPM
#> glue 1.7.0 2024-01-09 [2] RSPM
#> gtable 0.3.5 2024-04-22 [2] RSPM
#> hyperSpec * 0.200.0.9000 2024-05-27 [1] local
#> hySpc.testthat 0.2.1 2020-06-24 [2] RSPM
#> interp 1.1-6 2024-01-26 [2] RSPM
#> isoband 0.2.7 2022-12-20 [2] RSPM
#> jpeg 0.1-10 2022-11-29 [2] RSPM
#> jsonlite 1.8.8 2023-12-04 [2] RSPM
#> labeling 0.4.3 2023-08-29 [2] RSPM
#> lattice * 0.22-6 2024-03-20 [4] CRAN (R 4.4.0)
#> latticeExtra 0.6-30 2022-07-04 [2] RSPM
#> lazyeval 0.2.2 2019-03-15 [2] RSPM
#> lifecycle 1.0.4 2023-11-07 [2] RSPM
#> magrittr 2.0.3 2022-03-30 [2] RSPM
#> MASS 7.3-60.2 2024-05-25 [4] local
#> Matrix 1.7-0 2024-03-22 [4] CRAN (R 4.4.0)
#> mgcv 1.9-1 2023-12-21 [4] CRAN (R 4.4.0)
#> munsell 0.5.1 2024-04-01 [2] RSPM
#> nlme 3.1-164 2023-11-27 [4] CRAN (R 4.4.0)
#> pillar 1.9.0 2023-03-22 [2] RSPM
#> pkgbuild 1.4.4 2024-03-17 [2] RSPM
#> pkgconfig 2.0.3 2019-09-22 [2] RSPM
#> pkgload 1.3.4 2024-01-16 [2] RSPM
#> png 0.1-8 2022-11-29 [2] RSPM
#> praise 1.0.0 2015-08-11 [2] RSPM
#> processx 3.8.4 2024-03-16 [2] RSPM
#> ps 1.7.6 2024-01-18 [2] RSPM
#> R6 2.5.1 2021-08-19 [2] RSPM
#> RColorBrewer 1.1-3 2022-04-03 [2] RSPM
#> Rcpp 1.0.12 2024-01-09 [2] RSPM
#> R RcppEigen <NA> <NA> [?] <NA>
#> rematch2 2.1.2 2020-05-01 [2] RSPM
#> rlang 1.1.3 2024-01-10 [2] RSPM
#> rprojroot 2.0.4 2023-11-05 [2] RSPM
#> scales 1.3.0 2023-11-28 [2] RSPM
#> testthat 3.2.1.1 2024-04-14 [2] RSPM
#> tibble 3.2.1 2023-03-20 [2] RSPM
#> tidyselect 1.2.1 2024-03-11 [2] RSPM
#> utf8 1.2.4 2023-10-22 [2] RSPM
#> vctrs 0.6.5 2023-12-01 [2] RSPM
#> viridisLite 0.4.2 2023-05-02 [2] RSPM
#> waldo 0.5.2 2023-11-02 [2] RSPM
#> withr 3.0.0 2024-01-16 [2] RSPM
#> xml2 1.3.6 2023-12-04 [2] RSPM
#>
#> [1] /tmp/RtmpxUEHmP/temp_libpath17ce4b3a89e0
#> [2] /home/runner/work/_temp/Library
#> [3] /opt/R/4.4.0/lib/R/site-library
#> [4] /opt/R/4.4.0/lib/R/library
#>
#> R ── Package was removed from disk.
#>
#> ──────────────────────────────────────────────────────────────────────────────────────────────────References
Formulas are combined to a list by
c().↩︎Function
sweep()cannot be used here, and while there is the possibility to usesapply()ormapply(), they are not faster than the for loop in this case. Make sure to work on a copy of the spectra matrix, as that is much faster than row-wise extracting and changing the spectra by[[and[[<-.↩︎